El síndrome de Morris, también llamado síndrome de insensibilidad a los andrógenos (SIA) o feminización testicular, es una condición genética que afecta al desarrollo sexual. Los individuos que la padecen genéticamente son de sexo masculino, es decir, poseen un cromosoma X y otro Y en cada célula. Sin embargo, la forma corporal no coincide con la de dicho sexo.
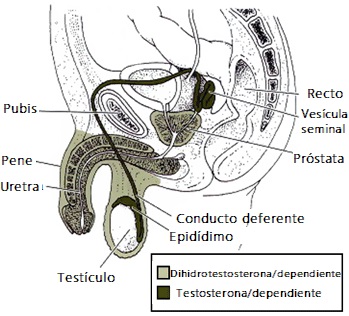
Para que se desarrolle un fenotipo masculino, no solo deben existir en ciertos niveles de hormonas masculinas (testosterona) en la sangre; también es necesario que funcionen adecuadamente los receptores androgénicos que las capten.

Lo que ocurre en este síndrome, es que existe un déficit en dichos receptores y por eso los tejidos del organismo no absorben la testosterona suficiente como para desarrollar una forma masculina.
Así, estos individuos nacen con aparentes genitales femeninos y suelen criarse como niñas. Cuando llegan a la pubertad se desarrollan caracteres secundarios femeninos (ensanchamiento de caderas, voz aguda, aumento de grasa) y las mamas. Sin embargo, se percatan de que no aparece la menstruación, ya que no poseen útero. Además, presentan escasez de vello de las axilas y en el pubis (o está ausente).
[toc]
Descubrimiento
El síndrome de Morris se descubrió en 1953 por el científico y ginecólogo John McLean Morris (de ahí su nombre). Después de observar 82 casos (dos eran pacientes propios), describió el “síndrome de feminización testicular”.
Morris pensaba que se debía a que los testículos de esos pacientes producían una hormona que tenía un efecto feminizante, sin embargo, ahora se sabe que es por la falta de acción de los andrógenos en el organismo.
Cuando no se absorbe la testosterona necesaria, el cuerpo tiende a desarrollarse con caracteres femeninos. No importa que se aumenten los niveles de testosterona, el problema reside en el cuerpo no la capta. Por eso actualmente se utiliza más el término de “resistencia a los andrógenos”.
También podemos encontrar el síndrome de Morris conceptualizado como pseudohermafroditismo masculino.
Prevalencia del síndrome de Morris
Según Borrego López, Varona Sánchez, Areces Delgado y Formoso Martín (2012); se estima que el síndrome de Morris se puede presentar en uno de cada 20000 a 64000 recién nacidos varones. Incluso la cifra podría ser mayor si se cuentan los casos aún no diagnosticados o que no solicitan asistencia médica.
El síndrome de Morris es considerado la tercera causa de amenorrea después de la disgenesia gonadal y de la ausencia de la vagina al nacer.
Tipos

No existe un grado único de insensibilidad a los andrógenos, sino que las características del síndrome dependen del nivel de déficit de los receptores androgénicos.
Así, pueden existir menos receptores de dihidrotestosterona de lo habitual y recibir menos cantidad de testosterona de la necesaria, o bien, puede haber casos en los que la deficiencia de receptores sea total.
Los tres tipos clásicos de insensibilidad a los andrógenos (SIA) son:
– Síndrome de insensibilidad a los andrógenos leve: genitales externos masculinos.
– Síndrome de insensibilidad a los andrógenos parcial: genitales parcialmente masculinizados.
– Síndrome de insensibilidad a los andrógenos completa: genitales femeninos.
El síndrome de Morris se enmarca en este último, ya que existe una resistencia a los andrógenos completa en la que los pacientes nacen con genitales externos femeninos.
En las formas incompletas, pueden aparecer diferentes niveles de rasgos masculinos y femeninos como clitoromegalia (clítoris más grande lo normal), o cierre parcial de la vagina externa.
Características y síntomas

Los individuos con el síndrome de Morris no van a manifestar síntomas en la infancia. De hecho, la mayoría recibe el diagnóstico cuando acude al especialista con el motivo de que no aparece la menstruación.

Las características que suelen presentar son las siguientes:
– Cariotipo 46 XY, que es el asociado al sexo masculino.
– Los genitales externos tienen apariencia femenina, aunque con hipoplasia de los labios mayores y menores. Eso significa que los labios no están completamente desarrollados, siendo de menor tamaño.
– A pesar de tener genitales externos normales, la vagina posee poca profundidad y termina en un fondo de saco ciego. Es decir, no está conectada al útero porque lo más habitual es que no se haya formado.
– A veces no presentan ovarios o éstos están atrofiados.
– Suelen tener testículos no descendidos que se encuentran en la región inguinal, en el abdomen o labios mayores. A veces los testículos se encuentran en el interior de una hernia inguinal que puede palparse en el examen físico.
Estos testículos son normales antes de llegar a la pubertad, pero después de ésta los túbulos seminíferos son más pequeños y no se produce la espermatogénesis.
– En la pubertad, se desarrollan los caracteres sexuales femeninos secundarios normales alcanzando total apariencia de mujer. Eso se debe a la acción del estradiol, una hormona sexual femenina que se produce en diversas partes del organismo.
Una característica distintiva del síndrome es que no presentan vello en las axilas ni en el pubis, o éste es muy escaso.
– Ausencia de menarquía (la primera menstruación).
– Los niveles de testosterona en sangre son típicos masculinos, pero al no existir una correcta función de los receptores androgénicos, las hormonas masculinas no pueden ejercer su labor.
– Como es lógico, esta enfermedad provoca infertilidad.
– Si no se interviene, son frecuentes las dificultades en las relaciones sexuales como problemas para llevar a cabo la penetración y dispareunia (dolor).
– Se ha encontrado en estos pacientes una disminución de la densidad ósea, que puede ser por la influencia de los andrógenos.
– Si no se extirpan los testículos, existe mayor riesgo de tumores malignos en las células germinales a medida que aumenta la edad. En un estudio ha sido estimado el riesgo en un 3,6% a los 25 años, y del 33% a los 50 años (Manuel, Katayama & Jones, 1976).
Causas
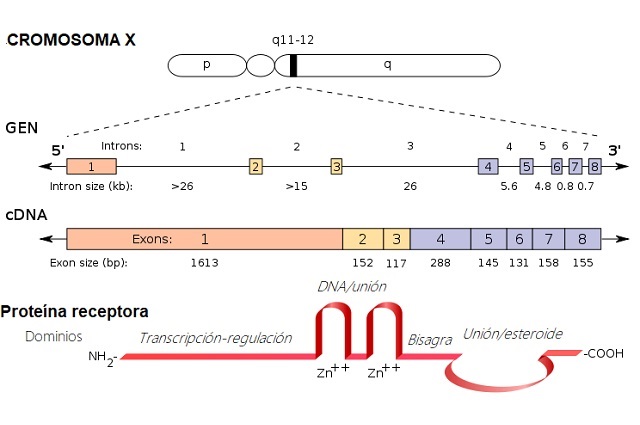
El síndrome de Morris es una condición hereditaria, con patrón recesivo ligado al cromosoma X. Esto significa que el gen mutado que provoca el síndrome se encuentra en el cromosoma X.
Aparece con más frecuencia en varones que en mujeres, ya que las mujeres necesitan mutaciones en ambos cromosomas (XX) para presentar el trastorno. En cambio, los hombres pueden desarrollarlo con una mutación en su cromosoma X (solo tienen uno).
Así, las mujeres pueden ser portadoras del gen mutado, pero no presentar el síndrome. De hecho, parece que aproximadamente dos tercios de todos los casos de resistencia a los andrógenos se heredan de madres que poseen una copia alterada del gen en uno de sus dos cromosomas X.
Los otros casos se deben a una nueva mutación que parece darse en el óvulo materno en el momento de la concepción o durante el desarrollo del feto (Genetics Home Reference, 2016).
Las mutaciones de este síndrome se localizan en el gen AR, que se encarga de enviar instrucciones para el desarrollo de proteínas AR (Receptoras de Andrógenos). Estas son las que median los efectos de los andrógenos en el organismo.
Los receptores captan las hormonas sexuales masculinas como la testosterona, enviándolas a las distintas células para que se produzca un desarrollo masculino normal.
Cuando este gen se altera, como ocurre en el síndrome de Morris, se pueden producir déficits tanto cuantitativos (cantidad de receptores) como cualitativos (receptores anómalos o que no funcionan bien) de los receptores androgénicos.
De esta forma, las células no responden a los andrógenos, es decir, las hormonas masculinas no hacen efecto. Por tanto, se impide el desarrollo del pene y otras características típicas del varón, y se da paso a un desarrollo femenino.
En concreto, la testosterona que existe en estos individuos se aromatiza (se transforma por la enzima aromatasa) en estrógeno, una hormona sexual que es la causante de la apariencia femenina en el síndrome de Morris.
Algunos rasgos de los varones se llegan a desarrollar, porque no son dependientes de los andrógenos. Por ejemplo, los testículos se forman debido al gen SRY presente en el cromosoma Y.
Diagnóstico
El diagnóstico del síndrome de Morris suele realizarse después de la pubertad, pues estos pacientes no suelen notar ningún síntoma antes de esta. No obstante, es un síndrome difícil de diagnosticar, ya que la apariencia es totalmente femenina y hasta que no se realiza un escáner del área pélvica o un estudio cromosómico no se detecta el problema.
Si se sospecha de la existencia del síndrome de Morris, el especialista llevará a cabo un diagnóstico basado en:
– Historia clínica completa del paciente, siendo importante que no haya presentado la menstruación.
– Exploración física que puede estar basada en la Escala de Tanner, que es aquella que refleja el nivel de maduración sexual. En este síndrome debería ser normal en las mamas, pero menor en los genitales y vello de las axilas y del pubis.
También puede utilizarse la Escala de Quigley, que mide el grado de masculinidad o feminidad de los genitales. Gracias a este índice puede distinguirse también entre los distintos tipos de insensibilidad a los andrógenos.
– Ultrasonido ginecológico: a través de ondas sonoras se obtienen imágenes de los genitales internos. Frecuentemente no se observa útero ni ovarios, pero si pueden estar presentes testículos en alguna zona cercana. La vagina suele poseer una longitud más reducida de lo normal.
– Estudios hormonales: a través de un análisis de sangre es conveniente explorar los niveles de testosterona (en el síndrome de Morris son elevados y similares a los niveles masculinos), Las Hormonas Estimuladoras del Folículo (FSH), las hormonas luteinizantes (LH) o el estradiol (E2).
– Estudio cromosómico: se pueden realizar a través de una muestra de sangre, biopsia de piel o cualquier otra muestra de tejido. En este síndrome el resultado debería ser un cariotipo 46 XY.
En la historia ha habido conflictos a la hora de decidir cuándo y cómo revelar un diagnóstico de Síndrome de Morris al afectado. En la antigüedad muchas veces se ocultaba por médicos y familiares, pero evidentemente esto tiene un impacto aún más negativo para la persona.
A pesar del dilema que genera, hay que intentar que el paciente reciba la información en un entorno empático y relajado, respondiendo a todas sus inquietudes.
Tratamiento
Actualmente no existe un método para corregir el déficit de los receptores de andrógenos presente en el síndrome de Morris. Pero hay otras intervenciones que pueden realizarse:
Terapia de dilatación
Antes de considerar la intervención quirúrgica, se intenta aumentar el tamaño de la vagina utilizando métodos de dilatación. Esto se recomienda llevar a cabo después de la pubertad.
Como la vagina es elástica, esta terapia consiste en la introducción y rotación de un objeto con forma fálica varias veces por semana durante unos minutos, siendo esto progresivo.
Gonadectomía
Es necesario extraer los testículos en los pacientes con el síndrome de Morris, ya que tienden a desarrollar tumores malignos (carcinomas) si no se retiran. Es determinante para un buen pronóstico que se extraigan lo antes posible.
Asistencia psicológica
Es esencial en estos pacientes que reciban tratamiento psicológico, ya que este síndrome puede provocar una importante insatisfacción con el propio cuerpo. A través de este tipo de intervención, la persona será capaz de aceptar su situación y llevar una vida lo más satisfactoria posible, evitando el aislamiento social.
Incluso se pueden trabajar los vínculos familiares, de forma que la familia apoye y contribuya al bienestar del paciente.
Suplementos
Para la disminución de la densidad ósea típica de estos pacientes, se aconsejan suplementos de calcio y de vitamina D. El ejercicio puede ser muy beneficioso también.
En casos más graves, se puede recomendar el uso de bifosfonatos, unos medicamentos que inhiben la resorción de los huesos.
Cirugía de construcción vaginal
Si los métodos de dilatación no han sido efectivos, puede ser una alternativa reconstruir una vagina funcional. El procedimiento se llama neovaginoplastia, y para la reconstrucción se utilizan injertos de piel del paciente del intestino o mucosa bucal.
Después de la cirugía, también serán necesarios los métodos de dilatación.
Reemplazo hormonal
Se ha intentado administrar estrógeno a estos pacientes para paliar la falta de densidad ósea, pero parece que esto no tiene el efecto deseado en todo el mundo.
Por otro lado, se ha administrado andrógenos después de la extirpación de los testículos (ya que se produce una bajada importante en el nivel de éstos). Aparentemente, los andrógenos mantienen una sensación de bienestar en las pacientes.
Referencias
- Borrego López, J.A., Varona Sánchez, J.A., Areces Delgado, G., & Formoso Martín, L. E. (2012). Síndrome de Morris. Revista Cubana de Obstetricia y Ginecología, 38(3), 415-423. Recuperado el 14 de octubre de 2016.
- Quigley C.A., De Bellis A., Marschke K.B., el-Awady M.K., Wilson E.M., French F.S. (1995). Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr. Rev. 16(3): 271–321.
- Manuel M., Katayama P.K., & Jones H.W. (1976). The age of occurrence of gonadal tumors in intersex patients with a Y chromosome. Am. J. Obstet. Gynecol. 124(3): 293–300.
- Hughes I.A., Deeb A. (2006). Androgen resistance. Best Pract. Res. Clin. Endocrinol. Metab. 20 (4): 577–98.
- Gottlieb B., Beitel L.K., Trifiro M.A. (1999). Androgen Insensitivity Syndrome. In: Pagon R.A., Adam M.P., Ardinger H.H., et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
- ¿De qué clases de pruebas se dispone para determinar la existencia de un defecto genético congénito en un niño? (s.f.). Recuperado el 14 de octubre de 2016, de University of Utah, Health care.
- Androgen insensitivity syndrome. (s.f.). Recuperado el 14 de octubre de 2016, de Wikipedia.
- Androgen insensitivity syndrome. (s.f.). Recuperado el 14 de octubre de 2016, de Medline Plus.
- Androgen insensitivity syndrome. (11 de octubre de 2016). Obtenido de Genetics Home Reference.
- Complete androgen insensitivity syndrome. (s.f.). Recuperado el 14 de octubre de 2016, de Wikipedia.